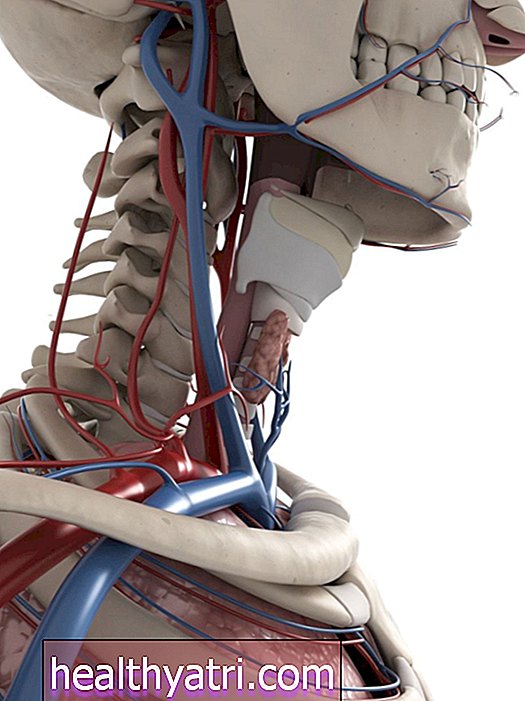
Spinal musküler atrofi (SMA), omurilikten dallanan kontrol sinirlerini istemli kaslar üzerinde etkileyen nadir bir genetik hastalıktır. SMA esas olarak çocukları etkiler.
SMA'lı bir çocuk nefes alma, emme ve yutma gibi önemli işlevlerde bozulma yaşayacaktır. Bu tür bozukluklardan ek koşullar gelişebilir. Örneğin, zayıf sırt kasları nedeniyle anormal omurga eğrileri gelişebilir ve akciğerleri sıkıştırarak solunum sürecini daha da karmaşık hale getirir.
Beslenme tüplerinin gelişinden önce, yutmanın kesilmesi, genellikle SMA tip 1 (en şiddetli tür) vakalarında ölüme neden oluyordu. Artık SMA'lı çocukların hayatta (ve en azından geçmiş yıllara göre rahat) kalmasına yardımcı olacak birçok yardımcı cihaz var.
Bununla birlikte, riskler hala mevcuttur. Biri boğuluyor. Boğulma mümkündür çünkü SMA'lı bir çocuğun zayıf yutma ve çiğneme kasları vardır. Diğer bir risk, yiyeceklerin aspirasyonu veya solunmasıdır. Aspirasyon hava yolunu tıkayabileceği gibi bir enfeksiyon kaynağı da olabilir.
SMA, özellikle türe göre değişecek birçok şekilde kendini gösterir. Tüm SMA türlerinde kas güçsüzlüğü, zayıflama ve atrofinin yanı sıra kas koordinasyon problemleri bekleyebilirsiniz. Bunun nedeni, hastalığın doğasında yatmaktadır - SMA, istemli kasların sinir kontrolünü etkiler.
SMA'nın tedavisi yoktur. En umut verici prognoz erken teşhisle ortaya çıkar. Tıptaki gelişmeler, SMA ile ilişkili komplikasyonların yönetilmesine yardımcı olabilir.
Spinal Musküler Atrofi Türleri
Spinal musküler atrofi 6.000 yenidoğandan 1'ini etkiler. 2 yaşın altındaki çocuklarda önde gelen genetik ölüm nedenidir. SMA kimi etkilediği konusunda ayrımcı değildir.
SMN proteininde görülen arıza derecesine bağlı olarak birkaç SMA türü vardır. Diğer genetik problemlerle ilgili bazı SMA çeşitleri de vardır.
SMA, semptomların başlangıç yaşına ve şiddetine göre kategorize edilir. Şiddet derecesi, motor nöronlardaki protein eksikliği miktarı ve (erken) başlangıç yaşı, birbirleriyle korelasyon gösterme eğilimindedir. SMA'da duyuların ve zihnin gelişimi tamamen normaldir.
Tür 1
Tip 1 SMA, 2 yaşın altındaki çocukları etkileyen en şiddetli olanıdır. Tip 1 SMA tanısı genellikle yaşamın ilk altı ayında konur.
Tip 1 SMA'lı bebekler, emme, yutma, dönme, oturma ve emekleme gibi normal motor gelişim başarılarına asla ulaşamazlar. SMA tip 1'li çocuklar, genellikle ilişkili solunum problemleri nedeniyle 2 yaşından önce ölme eğilimindedir.
SMA tip 1'e sahip bebekler gevşek, hareketsiz ve hatta sarkık olma eğilimindedir. Dilleri solucan gibi hareket eder ve oturma pozisyonuna getirildiğinde başlarını dik tutamazlar.
Ayrıca skolyoz gibi göze çarpan deformitelere sahip olabilirler ve özellikle omurganın yakınında bulunan proksimal kaslarda kas güçsüzlüğüne sahip olabilirler.
Tip 2
Ara SMA olarak da adlandırılan SMA tip 2, en yaygın SMA türüdür. Solunum yolu enfeksiyonu, tip 2'de en yaygın ölüm nedenidir. Bununla birlikte, tip 2'li çocuklar normal bir yaşam süresine sahip olabilir.
SMA tip 2, 6 ila 18 ay arasında veya çocuğun desteksiz oturabileceğini gösterdikten sonra (oturma pozisyonuna getirildikten sonra) başlar. Tip 2 semptomları arasında deformite, motor gecikme, genişlemiş baldır kasları ve parmaklarda titreme bulunur.
Omurgaya en yakın olan proksimal kaslar önce güçsüzlükten etkilenir; bacaklar kollardan önce zayıflayacak. Tip 2 SMA hastası çocuklar asla yardım almadan yürüyemezler. İyi haber şu ki, SMA'lı bir çocuk büyük olasılıkla kollarıyla ve elleriyle klavye kullanmak, beslemek gibi görevleri yerine getirebilecek.
SMA tip 2'li çocukların çok zeki olduğu görülmüştür. Fizik tedavi, yardımcı cihazlar ve elektrikli tekerlekli sandalyeler, onlar için anlamlı bir yaşama katkıda bulunmada uzun bir yol kat edebilir.
SMA Tip 2 ile ilgili iki ana sorun şunları içerir:
- Enfeksiyona neden olan zayıf solunum kasları
- Zayıf omurga kaslarına bağlı gelişen skolyoz ve / veya kifoz
Tip 3 ve 4
Hafif SMA olarak da bilinen SMA Tip 3, 18 ay sonra başlar. SMA tip 3 olan kişiler genellikle yardımcı cihazlara bağımlıdır ve yaşamları boyunca solunum ve omurga eğriliği riskleri açısından nerede olduklarını sürekli olarak izlemeleri gerekir. Hayatlarında bir süre yürümeyi bırakma eğilimindedirler. Yürümeyi bıraktıkları zaman ergenlik ve 40'lı yaşları arasında değişir.
Tip 3 SMA'lı çocuklar hareket edebilir ve yürüyebilirken, kas güçsüzlüğü ve proksimal kaslarda, yani omurgaya en yakın kaslarda zayıflık vardır.
Dördüncü tip SMA, yetişkin başlangıçlı SMA vardır. Tip 4 genellikle kişi 30'lu yaşlarında olduğunda ortaya çıkar. Tahmin edebileceğiniz gibi, SMA tip 4, bu hastalığın şiddetinin sürekliliği üzerindeki en hafif formdur. Tip 4'ün semptomları, tip 3'ün semptomlarına çok benzer.
Nedenleri
SMA, SMN (hayatta kalma motor nöronu) adı verilen bir kas proteinini kodlayan genin hatalı olduğu genetik bir bozukluktur. SMN proteininin arızalanması, SMA'da görülen sorunlara yol açar.
SMA, resesif bir modelde miras alınır. Bu, SMA'nın oluşması için, bir çocuğun kusurlu geni her iki ebeveynden de miras alması gerektiği ve bu nedenle, her iki ebeveynin de kusurlu genin taşıyıcısı olması gerektiği anlamına gelir. Yaklaşık 40 kişiden birinin bu genin taşıyıcısı olduğu tahmin edilmektedir. Her iki ebeveyn de taşıyıcıysa, onlardan doğan bir çocuğun SMA'ya sahip olma şansı dörtte birdir.
SMA'lı bazı insanlarda, diğer genler, hatalı SMN proteinleri üreten genleri kısmen telafi edebilir. Sonuç olarak, SMA'nın şiddeti kişiden kişiye biraz değişkendir.
Teşhis
Tanı almanın ilk adımı, ebeveynlerin veya bakıcıların, bu makalede belirtildiği üzere, çocuklarında SMA semptomlarını fark etmeleridir. Hekim, aile öyküsü ve fizik muayene dahil olmak üzere çocuğun ayrıntılı bir tıbbi öyküsünü almalıdır.
SMA'yı teşhis etmek için kullanılan birkaç test türü vardır:
- Kan testleri
- Kas biyopsisi
- Genetik testler
- EMG
Çocuklarda SMA testinin yanı sıra ebeveynlerin taşıyıcı durumu için test edilmesiyle ilgili birçok sorun üretilir. 1997'de, SMN1 geni için nicel PCR testi olarak adlandırılan bir DNA testi, ebeveynlerin SMA'ya neden olan mutant geni taşıyıp taşımadıklarını belirlemelerine yardımcı olmak için piyasaya çıktı.
Test, kan örneği alınarak yapılır. Genel popülasyonu test etmek çok zordur, bu nedenle ailelerinde SMA olan kişiler için ayrılmıştır. Amniyosentezler veya koryon villus örnekleri ile doğum öncesi test yapmak mümkündür.
Verywell / Lara AntalTedavi
SMA tedavisi, yaşam desteğine, bağımsızlığı teşvik etmeye ve / veya hastanın yaşam kalitesini iyileştirmeye odaklanır. Bakım ve tedavi yöntemlerinin örnekleri şunları içerir:
- Fizik Tedavi
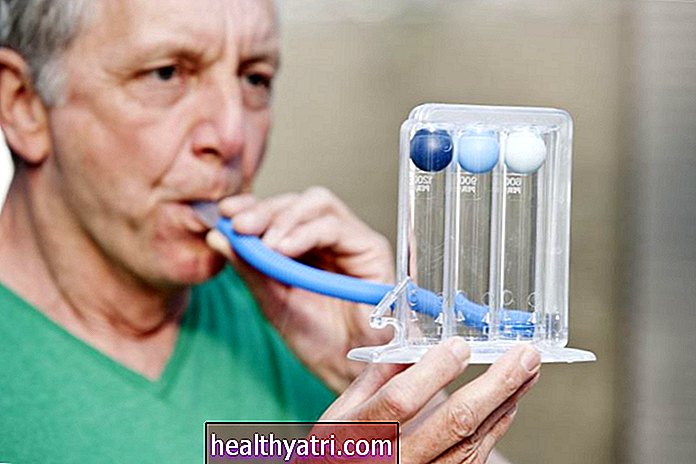
- Tekerlekli sandalyeler, solunum makineleri ve besleme tüpleri gibi yardımcı cihazların kullanımı. (SMA için pek çok yardımcı cihaz vardır. Bunu tedavi ekibinizle görüşmeniz en iyisidir.)
- Omurga deformitesi için cerrahi
Doktorlar, ailelerin multidisipliner bir yaklaşımla bir sağlık ekibi ile çalışmasını önermektedir. SMA hastası yaşamı boyunca oldukça sık tıbbi olarak değerlendirilmelidir. Aile üyeleri için genetik danışmanlık çok önemlidir.
Aktiviteden kaçınılmamalı, daha çok deformiteyi, kontraktürü ve sertliği önleyecek ve hareket açıklığını ve esnekliği koruyacak şekilde kullanılmalıdır. Bu nedenle tükenme noktasına kadar yapılmamalıdır. İyi beslenme, hastanın kaslarını da kullanmasını sağlayacaktır.
Spinal Musküler Atrofi (SMA) - Nadir Bir Kas Zayıflığı Türü